Hur kliniska prövningar av nya läkemedel genomförs. Experimentell studie och kliniska prövningar av läkemedel Mål för kliniska prövningar
Nu i världen finns det ett stort antal läkemedel för nästan alla befintliga sjukdomar. Utvecklingen av ett nytt läkemedel är inte bara tidskrävande utan också dyrt. Efter att medicinen har skapats är det nödvändigt att testa hur det verkar på människokroppen, hur effektivt det kommer att vara. För detta ändamål genomförs kliniska studier som vi kommer att prata om i vår artikel.
Kliniskt forskningskoncept
Någon forskning mediciner är helt enkelt nödvändiga, som ett av stadierna i utvecklingen av ett nytt läkemedel eller för att utöka indikationerna för användning av ett befintligt läkemedel. Först, efter att ha fått läkemedlet, utförs alla studier på mikrobiologiskt material och djur. Detta stadium kallas också preklinisk forskning. De görs för att få bevis på läkemedlets effektivitet.
Men djur skiljer sig från människor, så hur experimentella möss reagerar på läkemedlet betyder inte att man får samma reaktion hos människor.
Om vi \u200b\u200bger en definition av vad kliniska prövningar är, kan vi säga att detta är ett system för att använda olika metoder för att ta reda på säkerheten och effektiviteten hos ett läkemedel för människor. Under studiet av läkemedlet klargörs alla nyanser:
- Farmakologiska effekter på kroppen.
- Sughastighet.
- Läkemedlets biotillgänglighet.
- Uttagsperiod.
- Funktioner av ämnesomsättningen.
- Interaktion med andra läkemedel.
- Säkerhet för människor.
- Manifestation av biverkningar.
Laboratorieforskning börjar efter bedömning av sponsor eller kund, som inte bara ansvarar för organisationen utan också för kontrollen och finansieringen av denna procedur. Oftast är en sådan person det läkemedelsföretagsom utvecklade detta läkemedel.
Alla resultat från kliniska prövningar, deras kurs bör beskrivas i detalj i protokollet.
Forskningsstatistik
Studien av läkemedel utförs över hela världen; detta är ett obligatoriskt steg före registrering av ett läkemedel och dess massutsläpp för medicinskt bruk. De medel som inte har testats kan inte registreras och införas på läkemedelsmarknaden.
Enligt en av de amerikanska läkemedelsproducentorganisationerna, av 10 000 undersökningsdroger, når endast 250 det prekliniska stadiet, som ett resultat kommer kliniska prövningar endast att genomföras för cirka 5 läkemedel och 1 kommer att nå massproduktion och registrering. Det här är statistiken.
Laborationsforskningsmål
Forskning på något läkemedel har flera mål:
- Bestäm hur säkert detta läkemedel är för människor. Hur kroppen tål det. För att göra detta, hitta volontärer som går med på att delta i studien.
- Under studiens gång väljs de optimala doserna och behandlingsregimerna för att uppnå maximal effekt.
- Att fastställa graden av läkemedelssäkerhet och dess effektivitet för patienter med en specifik diagnos.
- Studie av oönskade biverkningar.
- Överväg att utöka droganvändningen.
 Ganska ofta utförs kliniska prövningar samtidigt i förhållande till två eller till och med tre läkemedel, så att deras effektivitet och säkerhet kan jämföras.
Ganska ofta utförs kliniska prövningar samtidigt i förhållande till två eller till och med tre läkemedel, så att deras effektivitet och säkerhet kan jämföras.
Forskningsklassificering
En fråga som klassificeringen av läkemedelsstudier kan hanteras från olika vinklar. Beroende på faktor kan olika typer av forskning vara olika. Här är några sätt att klassificera:
- Genom graden av störning i patienthanteringens taktik.
- Forskning kan skilja sig åt i målen.
Dessutom finns det också typer laboratorieforskning... Låt oss undersöka denna fråga mer detaljerat.
Varianter av patientinterventionsstudier
Om vi \u200b\u200böverväger klassificeringen i termer av intervention vid standardbehandling är studierna uppdelade i:
- Observational. Under en sådan studie inträffar ingen störning, information samlas in och den naturliga förloppet för alla processer observeras.
- Forskning utan intervention eller icke-intervention. I det här fallet ordineras läkemedlet som vanligt. Forskningsprotokollet avgör inte i förväg frågan om att tillskriva patienten någon behandlingstaktik. Förskrivning skiljer sig tydligt från patientregistrering. Patienten genomgår inga diagnostiska procedurer, data analyseras med epidemiologiska metoder.
- Interventionell forskning. Det utförs när det är nödvändigt att studera fortfarande oregistrerade läkemedel eller att ta reda på nya riktningar vid användning av kända läkemedel.

Klassificeringskriterium - forskningsmål
Beroende på syftet kan generella kliniska prövningar vara:
- Förebyggande. De utförs för att hitta de bästa sätten att förebygga sjukdomar hos en person som han inte tidigare har lidit eller för att förhindra återfall. Vanligtvis studeras vacciner, vitaminpreparat på detta sätt.
- Screeningtest gör att du kan hitta den bästa metoden för att upptäcka sjukdomar.
- Diagnostiska studier utförs för att hitta mer effektiva sätt och metoder för att diagnostisera sjukdomen.
- Terapeutisk forskning ger en möjlighet att studera läkemedlets effektivitet och säkerhet och terapimetoder.

- Livskvalitetsforskning bedrivs för att förstå hur livskvaliteten för människor med vissa sjukdomar kan förbättras.
- Utökade åtkomstprogram involverar användning av ett experimentellt läkemedel hos patienter med livshotande sjukdomar. Normalt kan sådana läkemedel inte ingå i laboratorietester.
Forskningstyper
Förutom typerna av forskning finns det också typer som du behöver bekanta dig med:
- En pilotstudie genomförs för att samla in nödvändiga data för nästa steg i läkemedelsstudien.
- Randomisering förutsätter att patienter slumpmässigt tilldelas grupper, de har möjlighet att ta emot både studieläkemedlet och kontrollläkemedlet.

- En kontrollerad studie av ett läkemedel undersöker ett läkemedel vars säkerhet ännu inte är känt. Det jämförs med ett redan välundersökt och välkänt läkemedel.
- En okontrollerad studie innebär inte en kontrollgrupp för patienter.
- En parallell studie utförs på flera grupper av patienter samtidigt som får studieläkemedlet.
- I crossover-studier får varje patient båda läkemedlen som jämförs.
- Om studien är öppen känner alla deltagare läkemedlet som patienten tar.
- Blindt eller förklädt lärande involverar två parter som inte är medvetna om patientgruppering.
- En prospektiv studie genomförs med patienter som tilldelats grupper oavsett om de får studieläkemedlet innan resultaten uppstår.
- I efterhand beaktas resultaten av redan genomförda studier.
- Ett kliniskt forskningscenter kan vara involverat en eller flera, beroende på detta finns det enstaka eller multicenterstudier.
- I en parallell studie jämförs resultaten från flera grupper av försökspersoner samtidigt, varav en är en kontroll, och två eller flera andra får studieläkemedlet.
- Liknande fallstudier innebär att man jämför patienter med en specifik sjukdom med de utan en sådan sjukdom för att identifiera ett samband mellan utfall och tidigare exponering för vissa faktorer.
Forskningsstadier
Efter framställningen av ett läkemedel måste han genomgå alla studier och de börjar med prekliniska. De utförs på djur och hjälper läkemedelsföretaget att förstå om läkemedlet är värt att undersöka ytterligare. 
Hos människor kommer ett läkemedel att testas först efter att det har bevisats att det kan användas för att behandla ett visst tillstånd och det inte är farligt.
Utvecklingsprocessen för alla läkemedel består av fyra faser, var och en är en separat studie. Efter tre framgångsrika steg får läkemedlet ett registreringsbevis och det fjärde är redan en studie efter registrering.
Fas ett
I det första steget reduceras en klinisk studie av läkemedlet till rekrytering av volontärer från 20 till 100 personer. Om ett läkemedel som är för giftigt undersöks, till exempel för behandling av onkologi, väljs patienter som lider av denna sjukdom.
Oftast genomförs den första fasen av studien i speciella institutioner där det finns kompetent och utbildad personal. Under detta skede måste du ta reda på:
- Hur läkemedlet tolereras av människor.
- Farmakologiska egenskaper.
- Period för absorption och utsöndring från kroppen.
- Preliminär bedömning av säkerheten vid mottagningen.
I den första fasen används olika typer av forskning:
- Användningen av enstaka ökande doser av läkemedlet. Den första gruppen försökspersoner injiceras med en viss dos av läkemedlet, om det tolereras väl, ökas nästa grupp doseringen. Detta görs tills de avsedda säkerhetsnivåerna nås eller börjar manifestera. bieffekter.
- Studier av flera eskalerande doser. En grupp volontärer får ett litet läkemedel flera gånger, efter varje kvitto tas tester och läkemedlets beteende i kroppen utvärderas. I nästa grupp administreras en ökad dos upprepade gånger och så vidare upp till en viss nivå.
Andra fasen av forskningen
Efter att läkemedlets säkerhet tidigare har utvärderats fortsätter kliniska forskningsmetoder till nästa steg. För detta rekryteras redan en grupp på 50-100 personer.
Huvudmålet i detta skede av läkemedelsstudien är att bestämma erforderlig dosering och behandlingsregim. Mängden läkemedel som ges till patienter i denna fas är något lägre än de högsta doserna som ges till försökspersoner i den första fasen.
I detta skede finns det definitivt en kontrollgrupp. Effekten av ett läkemedel jämförs antingen med placebo eller med ett annat läkemedel som har visat sig vara mycket effektivt vid behandling av denna sjukdom.
Fas 3-forskning
Efter de två första faserna fortsätter läkemedlet att undersökas i den tredje fasen. En stor grupp människor upp till 3000 personer deltar. Syftet med detta steg är att bekräfta läkemedlets effektivitet och säkerhet.
Även i detta skede studeras beroendet av resultatet på läkemedlets dos.
Efter att läkemedlet i detta skede har bekräftat dess säkerhet och effektivitet utarbetas ett registreringsunderlag. Den innehåller information om resultaten av studien, läkemedlets sammansättning, hållbarhet och lagringsförhållanden.
Fas 4
Detta stadium kallas redan forskning efter registrering. Fasens huvuduppgift är att samla in så mycket information som möjligt om resultaten av långtidsanvändning av läkemedlet av ett stort antal människor.
Frågan om hur läkemedel interagerar med andra medel studeras också, vilken är den mest optimala behandlingsperioden, hur läkemedlet påverkar patienter i olika åldrar.
Forskningsprotokoll
Varje forskningsprotokoll bör innehålla följande information:
- Syftet med att studera medicinen.
- De uppgifter som forskarna ställde för sig själva.
- Studera design.
- Studiemetoder.
- Statistiska frågor.
- Organisationen av själva studien.

Utvecklingen av protokollet börjar redan innan all forskning börjar. Ibland kan denna procedur ta flera år.
När studien är klar är protokollet det dokument som revisorer och inspektörer kan verifiera det mot.
Nyligen har olika metoder för klinisk laboratorieforskning använts allt oftare. Detta beror på att principerna för evidensbaserad medicin införs aktivt i vården. En av dem är att fatta beslut för patientens behandling baserat på bevisade vetenskapliga data, och det är omöjligt att få dem utan att genomföra en omfattande studie.
»» Nr 3 "99
Klinisk farmakologi
L.S. Strachunsky, M.M. Fjättrar
Smolensk State Medical Academy
En av huvudtyperna av kliniska prövningar är kliniska prövningar av läkemedel, vars principer diskuteras i denna artikel.
Läkare och patienter vill vara säker på att de ordinerade läkemedlen kommer att lindra symtomen eller läka patienten. De vill också att behandlingen ska vara säker. Det är därför det är nödvändigt att genomföra kliniska prövningar på människor. Kliniska prövningar är en nödvändig del av processen att utveckla något nytt läkemedel eller utvidga indikationerna för ett läkemedel som läkare redan känner till. Resultaten av kliniska prövningar överlämnas till officiella myndigheter. (I vårt land är detta Ryska federationens hälsovårdsministerium och den statliga farmakologiska kommittén och Institutet för preklinisk och klinisk expertis för läkemedel som är underordnat det.) Om studier har visat att läkemedlet är effektivt och säkert, ger Ryska federationens hälsovårdsministerium tillstånd att använda det.
Oumbärlighet för kliniska prövningar.
Kliniska prövningar kan inte ersättas med vävnad (in vitro) eller försöksdjur, inklusive primater. Organism hos försöksdjur skiljer sig från människors farmakokinetiska egenskaper (absorption, distribution, metabolism och utsöndring av läkemedlet) såväl som vad organ och system svarar på läkemedlet. Om ett läkemedel orsakar blodtrycksfall hos en kanin betyder det inte att det kommer att fungera i samma utsträckning hos människor. Dessutom är vissa sjukdomar unika för människor och kan inte simuleras i ett laboratoriedjur. Dessutom, även i studier på friska volontärer, är det svårt att på ett tillförlitligt sätt återge de effekter som ett läkemedel kommer att orsaka hos patienter.
Kliniska undersökningar är en oundviklig typ av vetenskaplig verksamhet, utan vilken det är omöjligt att få och välja nya, effektivare och säkrare läkemedel, liksom att "rena" medicin från föråldrade ineffektiva läkemedel. Nyligen har kliniska prövningar ökat i samband med införandet av principerna för evidensbaserad medicin i praktisk hälsovård. Chefen bland dem tar specifika kliniska beslut för att behandla en patient, inte så mycket på grundval av personlig erfarenhet eller expertutlåtande, baserat på starka vetenskapliga bevis som kan erhållas från väldesignade, kontrollerade kliniska prövningar.
Forskningssekvensen.
När man studerar ett nytt läkemedel observeras alltid forskningssekvensen: från celler och vävnader till djur, från djur till friska frivilliga, från ett litet antal friska frivilliga till sjuka.
Trots den otvivelaktiga begränsade information som erhållits i studier på försöksdjur testas läkemedlet på dem innan det först används på människor (prekliniska prövningar). Deras huvudsyfte är att få information om toxiciteten hos ett nytt läkemedel. Studera akut toxicitet med en enda dos och subakut toxicitet vid upprepad administrering av läkemedlet; undersöka mutagenicitet, effekter på reproduktions- och immunförsvaret.
Tabell 1. Faser av kliniska prövningar
| Fas | Typiskt antal patienter | Huvuduppgifter |
| Jag | 20-80 | Första användningen av läkemedlet hos människor, bedömning av toxicitet och säkerhet, bestämning av farmakokinetiska parametrar |
| II | 100-800 | Fastställande av effektivitet, bestämning av optimala doseringsregimer, säkerhetsbedömning |
| III | 1000-4000 | Bekräftelse av effekt- och säkerhetsdata, jämförande studier med standardläkemedel |
| IV | Tiotusentals | Ytterligare studier av effekt för att optimera läkemedelsanvändning, långsiktiga säkerhetsstudier, bedömning av sällsynta biverkningar |
Läkemedlets utvecklingsfaser
Vidare utförs kliniska prövningar, uppdelade i fyra faser. Tabell 1 och figuren visar deras huvudsakliga egenskaper. Som framgår av tabellen tillåter uppdelningen i faser att studera ett nytt läkemedel hos en person gradvis och sekventiellt. Först studeras det hos ett litet antal friska volontärer (fas I) - (endast vuxna kan vara volontärer) och sedan på ett ökande antal patienter (fas II-III). Det är oacceptabelt att "hoppa över" faserna i kliniska prövningar, studien fortsätter sekventiellt från fas I till IV. Målen och målen för de genomförda testerna bör förändras beroende på den information som erhållits under tidigare forskning. Kliniska studier kan avslutas i valfri fas i händelse av data om läkemedlets toxicitet.
Garantera patienters rättigheter och följa etiska standarder,
som är ett särskilt fall av respekt för mänskliga rättigheter, utgör hörnstenen i hela systemet för klinisk forskning. De regleras av internationella avtal (Helsingforsdeklarationen från World Medical Association) och den ryska federala lagen om läkemedel.
På lokal nivå är garantin för patienträttigheter etikkommittén, vars godkännande måste erhållas innan all forskning börjar. Den består av medicinska och vetenskapliga arbetare, advokater, präster osv. Vid sina möten överväger medlemmarna i etikkommittén information om läkemedlet, protokollet för den kliniska prövningen, texten till informerat samtycke och vetenskapliga biografier av forskarna när det gäller att bedöma risken för patienter, iaktta och garantera deras rättigheter ...
Frivilligt deltagande i klinisk forskning innebär att patienten endast kan delta i forskningen med fullt och medvetet frivilligt samtycke. Att få informerat samtycke från en sannolik patient är kanske en av de svåraste uppgifterna som utredaren står inför. Under alla omständigheter bör dock varje patient informeras fullständigt om konsekvenserna av deras deltagande i en klinisk prövning. Informerat skriftligt samtycke, på ett språk som är förståeligt för en lekman, anger målen för studien, fördelarna som patienten får av att delta i den, de kända biverkningarna som är förknippade med studieläkemedlet, villkoren för patientens försäkring etc.
Den utredande läkaren måste svara på alla patientens frågor. Patienten ska ges möjlighet att diskutera studien med familj och vänner. Enligt den federala lagen "On Medicines" måste en sådan samtycke ges av deras föräldrar när det gäller en klinisk prövning på barn. Det är förbjudet att utföra kliniska prövningar av läkemedel, inklusive vacciner och serum, på minderåriga utan föräldrar.
En av huvudaspekterna för att skydda patienternas rättigheter är att sekretessen för information om patienten bibehålls. Således kan patientens personuppgifter (efternamn, förnamn, patronym, hemvist) endast nås av personer som direkt deltar i studien. I all dokumentation anges endast patientens individuella nummer och initialer.
Enheten i det metodiska tillvägagångssättet.
Alla kliniska prövningar måste utföras enligt vissa regler. En studie som genomförts i Ryssland bör inte skilja sig från studier som genomförts i andra länder när det gäller metodiska metoder, oavsett om ett inhemskt eller utländskt läkemedel testas, studien sponsras av ett läkemedelsföretag eller en statlig organisation.
Sådana regler har redan skapats och kallas kvalitet klinisk praxis (PCC), som är en av de möjliga översättningarna av den engelska termen Good Clinical Practice (GCP).
PSC: s huvudregler
(GCP) är att skydda rättigheterna för patienter och friska frivilliga som deltar i en klinisk prövning och för att få tillförlitliga och reproducerbara data. Det senare uppnås genom att följa följande principer: 1) ansvarsfördelning mellan forskningsdeltagare; 2) deltagande av kvalificerade forskare; 3) närvaron av extern kontroll; 4) vetenskapliga synsättet att planera studien, registrera data, analysera och presentera dess resultat.
PCC-reglerna anger att vid genomförande av en klinisk prövning ska alla plikter och ansvar för genomförandet av enskilda delar av arbetet tydligt fördelas mellan alla deltagare i studien redan innan den börjar. Det finns tre huvudsakliga parter inblandade i studien: arrangören, forskaren och monitoren (en person eller en grupp människor som kontrollerar det direkta genomförandet av studien i kliniken).
Ansvaret för arrangörerna av studien.
Forskningsorganisatörerna (sponsorerna) kan vara läkemedelsföretag eller forskarna själva. Sponsorn ansvarar för att organisera och genomföra forskningen som helhet. För att göra detta måste han utveckla ett studieprotokoll, förse utredaren med studieläkemedlet, tillverkat och förpackat i enlighet med CCP-standarderna, och fullständig information om honom. Informationen bör innehålla data från alla prekliniska och tidigare kliniska prövningar, inklusive information om alla biverkningar. Försäkring för patienter och utredare är också sponsorens ansvar.
Forskarnas ansvar.
Forskare är främst ansvariga för klinikens etik och praktik och för patienternas hälsa och välbefinnande under studien. Kliniska prövningar kan endast utföras av läkare med lämpliga kvalifikationer och officiellt tillstånd att praktisera som läkare. Komponenterna i forskarnas utbildning är deras yrkesutbildning och special träning enligt kliniska prövningar och PCC-regler.
Forskare bör alltid vara beredda att genomföra kvalitetskontroller av sitt arbete. Kontrollerna är indelade i flera typer: övervakning, revision och inspektion. Monitorn kontrollerar regelbundet efterlevnaden av de etiska standarderna för studien och studieprotokollet, samt kvaliteten på att fylla i dokumentationen. Granskningen utförs vanligtvis bara en gång, i de viktigaste studierna. Syftet med revisionen är att verifiera överensstämmelse med CCP-reglerna, protokollet och lokal lagstiftning. Granskningens varaktighet beror på studiens komplexitet och kan ta flera dagar. Inspektionen strävar efter samma mål; den utförs av officiell kontroll och tillåtna organ.
Forskningsplanering.
Det är viktigt att forskningen planeras och genomförs i enlighet med de modernaste vetenskapliga standarderna. Formellt efterlevnad av CCP-reglerna garanterar inte att meningsfull information kommer att erhållas. För närvarande beaktas endast de resultat som erhållits i prospektiva, jämförande, randomiserade och helst dubbelblinda studier (tabell 2). Innan studien påbörjas måste ett protokoll (program) utvecklas, vilket är en skriftlig studieplan. Tabell 3 visar de avsnitt som måste återspeglas i protokollet.
Det finns inga studier utan fel, men reglerna för att genomföra kliniska prövningar bör aldrig brytas (tabell 4).
Tabell 2. Studieegenskaper
| Studie | Definition | mål |
| Blivande | Genomföra forskning enligt en förutvecklad plan | Ökad tillförlitlighet hos data, eftersom sannolikheten att den observerade effekten beror på en tillfällighet snarare än att det läkemedel som undersöks minskar. Kontroll över möjliga systematiska fel i analysen av resultaten |
| Jämförande | Jämförelse av effekterna i två grupper av patienter, en som fick studieläkemedlet och den andra som fick ett jämförande läkemedel eller placebo | Eliminera möjligheten att effekten beror på spontan sjukdom och / eller placeboeffekt |
| Slumpmässigt | Slumpmässigt tilldelning av patienter till studie- och kontrollgrupper | Eliminera eller minimera skillnader i baslinjeegenskaper mellan studiegrupper. Grund för korrekt tillämpning av de flesta statistiska tester |
| Dubbelblind | Varken patienten eller utredaren vet vilket läkemedel patienten får: undersökande eller kontrollerad | Eliminera fördomar vid utvärdering av effekten av undersökningsdroger |
Tabell 3. Huvudavsnitt i protokollet
Tabell 4. När du utför kliniska prövningar kan du inte:
- Genomför forskning utan ett noggrant utformat protokoll
- Starta forskning utan godkännande av dess material av en oberoende etikkommitté
- Inkludera en patient i en studie utan att få skriftligt informerat samtycke
- Kränka protokollkraven under studien:
- inkludera patienter som bryter mot kriterierna för inkludering och uteslutning;
- störa schemat för patientbesök;
- ändra läkemedelsregimen;
- ordinera förbjudna samtidigt läkemedel;
- utföra mätningar (undersökningar) med olika enheter, bryta mot undersökningsschemat
- Rapportera inte biverkningar
På tal om genomförande av kliniska prövningar inom barnläkemedel bör det noteras att kliniska prövningar av läkemedel hos minderåriga utförs i fall där undersökningsmedlet enbart är avsett för behandling av barnsjukdomar eller när syftet med kliniska prövningar är att erhålla data om den bästa dosen av läkemedlet för behandling av barn. Kliniska studier av ett läkemedel hos barn bör föregås av kliniska studier på vuxna och en grundlig analys av erhållna data. De resultat som erhållits hos vuxna är grunden för att planera forskning på barn.
I detta fall bör alltid komplexiteten i farmakokinetik, farmakodynamik och dosering av läkemedel hos barn beaktas. Farmakokinetiska studier av läkemedel bör utföras på barn i olika åldersgrupper med beaktande av de snabbt föränderliga processerna för absorption, distribution, metabolism och utsöndring av läkemedel, särskilt under den nyfödda perioden. Vid val av undersökningsmetoder och måttmätningar bör icke-invasiva metoder föredras, det är nödvändigt att begränsa frekvensen av blodprov hos barn och det totala antalet invasiva undersökningsmetoder.
Rättslig grund för kliniska prövningar.
Utförandet av kliniska prövningar i vårt land regleras av den federala lagen "On Medicines" daterad 22.06.1998, som har ett separat kapitel IX "Utveckling, prekliniska och kliniska prövningar av läkemedel". Enligt denna lag kan klinisk forskning endast utföras på kliniker som har lämpliga licenser. Licenser utfärdas endast till de kliniker som kan säkerställa genomförande av kliniska prövningar av läkemedel i enlighet med reglerna i PCC.
Federal Agency for Quality Control of Medicines utfärdar tillstånd för att genomföra dessa kliniska prövningar, varefter ett avtal ingås mellan kliniken där studien är planerad och arrangören av prövningen. I händelse av att betalning för testet tillhandahålls kan det göras av arrangören av studien endast via banköverföring, i enlighet med det avtal som ingåtts med kliniken.
I slutet av vårt sekel började alla inse kraften i moderna läkemedel, tack vare vilka inte bara rent medicinska (minskar patientens lidande, räddar eller förlänger livet) utan också sociala problem (förbättring av livskvaliteten) löses. Hundratals nya läkemedel godkänns för utbredd användning varje år. Utan kliniska prövningar är det omöjligt att utveckla utvecklingen av nya läkemedel. Men ingenting: varken en forskares intressen eller ett läkemedelsföretags intressen eller den kliniska farmakologins intressen i allmänhet borde vara högre än ett barns rättigheter och intressen som juridiskt sett kan vara föremål för forskning.
Litteratur
1. Helsingforsdeklarationen. Rekommendationer som guidar läkare i biomedicinsk forskning som involverar människor, The World Medical Association, 1964 (reviderad 1996).
2. Federal lag av 22.06.98. N86 FZ "On Medicines" (antagen av statsduman i Federal Assembly Ryska Federationen 06/05/98), Ryska federationens samlade lagstiftning, N26, 06/29/98, artikel 3006.
3. ICH Topic 6 - Guideline for Good Clinical Practice, Good Clinical Practice J., 1996, v. 3, No. 4 (Suppl.).
Mycket negativt? När allt kommer omkring hjälper vissa verktyg verkligen. Nackdel folkmedicin det faktum att effektiviteten hos de flesta av dem inte testas med rigorösa vetenskapliga metoder, så det finns alltid en stor risk för fel. Men det finns också effekten ” placebo»- självhypnos, när patienten övertygar sig om att medicinen verkligen hjälper, även om det kan vara vanligt kranvatten.
Förra gången pratade jag om vad kliniska prövningar av läkemedel är och idag kommer jag att fokusera på tekniken för att genomföra dem och utvärdera resultaten.
Vad är GCP
Det finns en internationell gMP-standard (god tillverkningssed) och för läkemedelskliniska prövningar, gCP-standard (bra klinisk praxis är bra klinisk praxis).
Varje patient som deltar i studien måste ge sitt skriftliga godkännande för behandling med en möjlig placebo. Får han betalt? Vanligtvis inte. Patienten får helt enkelt gratis behandling. Forskningsprotokollet för ett nytt läkemedel måste godkännas Etik kommitten varje medicinsk institution där testerna utförs. Varför är detta nödvändigt? Det finns en fin linje här. Läkaren har ingen rätt att använda placebo hos allvarligt sjuka patienter om det kan sluta tragiskt (till exempel när det gäller appendicit läkaren är skyldig att ordinera en operation till patienten, även om det inte fanns några jämförande kliniska studier om fördelarna med sådana operationer, men det fanns bara observationer av kirurger att patienter dog utan operation). Vid utveckling av hotande komplikationer ska placebo stoppas omedelbart. Om patienten slutar ta det föreskrivna läkemedlet när som helst, lämnar han studien.
För varje patient fylls en rapport i form av ett separat kort CRF (blankett för ärenderapport), inklusive originalet och två exemplar, varav en finns kvar i vårdcentralen och lagras i 15 år.
Varje forskare måste ge detaljerad information om sig själv och måste omedelbart informera det upphandlande företaget om allvarliga biverkningar som identifierats. Vissa studier har varit stannade före schematnär forskare fick starka bevis för negativa behandlingsresultat (till exempel en signifikant ökning av dödligheten i behandlingsgruppen). Men det händer också att kliniska prövningar avbryts tidigt om det finns en enorm fördel med ett nytt läkemedel eller behandling.
Slutpunkter
För att utvärdera resultaten av en studie måste du välja vissa parametrar som ska utvärderas. Parametrarna sorteras i minskande ordningsföljd (primära, sekundära och tertiära slutpunkter).
Primär ("solid") slutpunkter är parametrar relaterade till patienternas liv och utvecklingen av livshotande komplikationer. Organismen som helhet utvärderas. Exempel:
- allmän dödlighet,
- förekomsten av dödlig hjärtinfarkt, stroke, ventrikelflimmer etc.
Sekundära och tertiära punkter kallas också " mjuk"Och" surrogat».
Sekundär slutpunkter återspeglar tillståndet för ett eller två kroppssystem:
- förbättrad livskvalitet på grund av lindring av symtomen på sjukdomen (till exempel en minskning av frekvensen av anginaattacker),
- frekvens av icke-dödlig (icke-dödlig) hjärtinfarkt,
- minska förekomsten av icke-dödliga sjukdomar (till exempel förmaksflimmerparoxysm).
Tertiär slutpunkter återspeglar förändringar i enskilda parametrar, såsom kolesterolnivåer eller blodtryck.
När du utvärderar ett nytt läkemedel ska du alltid lita på det "Solid" (primär) slutpunkter. Att endast utvärdera mjuka punkter kan leda till allvarliga fel. Det är förmodligen varför poängen kallas surrogat? Exempel:
- hjärtglykosider vid kronisk hjärtsvikt ökar de kraften av hjärtmuskelkontraktioner (tertiär punkt), minskar frekvensen av sjukhusvistelser och förbättrar livskvaliteten (sekundära poäng), men leder inte till en minskning av total dödlighet (primär punkt) på grund av en ökad förekomst av dödlig arytmi (också en primär punkt);
- med aids vissa läkemedel som ökar nivåerna av T-hjälpar (tertiär slutpunkt) minskade inte dödligheten (primärt slutpunkt). För din information: T-hjälpare är en typ av lymfocyt som attackeras av HIV.
Mega-forskning
Ju mer kvalitativ forskning som utförs, desto mer tillförlitliga resultat.
Mega-forskning(från mega - enorm) - detta är studien av nya läkemedel för mer än 10 tusen patienter. I små grupper av patienter är resultaten inte så tillförlitliga, eftersom i små grupper:
- det är svårt att skilja ett positivt resultat från behandling från spontan remission av sjukdomen,
- det är svårt att uppnå gruppers homogenitet,
- det är svårt att identifiera små positiva förändringar i behandlingen och ytterligare prognoser,
- sällsynta biverkningar är svåra att upptäcka.
Ibland beror statistiskt tillförlitliga data från en megastudie om fördelarna med ett nytt läkemedel på närvaron av en liten grupp patienter som är mycket känsliga för behandling bland ett stort antal patienter. Resten av det nya läkemedlet ger inte mycket nytta. Sådana patienter som är mycket känsliga för behandling bör identifieras. det nya läkemedlet ger bara dem maximal nytta.
Diagram över en heterogen studiemodell
Meta-analys
Meta-analys (Grekisk. meta - genom) - kombinerar resultaten från flera kontrollerade studier om ett ämne. När antalet analyserade studier ökar kan nya positiva och negativa effekter av behandlingen hittas som inte sågs i enskilda studier. Metaanalyser (metaanalyser) är den absolut viktigaste och mest värdefulla eftersom forskare analyserar kvaliteten på många kliniska studier om ett givet ämne, en del av olika anledningar avvisas och slutsatser dras från resten.
Som du borde veta vid det här tillfället är det viktigt att först utvärdera de primära slutpunkterna när du läser resultaten av all forskning. Till exempel fann två metaanalyser en positiv antiarytmisk effekt lidokain med hjärtinfarkt, och en metaanalys var negativ. Vad ska jag tro? Rekommendera lidokain till alla med hjärtinfarkt? Nej, eftersom de två första metaanalyserna fokuserade på effekten av lidokain på arytmier (dvs att bedöma sekundära slutpunkter) och den tredje på effekten av lidokain på överlevnad i hjärtinfarkt (primär slutpunkt). Således undertryckte lidokain framgångsrikt arytmier, men ökade samtidigt dödligheten hos patienter.
Nackdelar med metaanalyser
Metaanalyser ersätter inte megastudier och kan i vissa fall till och med motsäga de senare. Metaanalyser kan vara föga övertygande i följande fall:
- om metaanalysen ger en generaliserad slutsats, även om studierna involverade en heterogen grupp patienter. Eller började behandlingen in olika termer och i olika doser läkemedel;
- om effektiviteten av behandlingen jämförs i vissa grupper med placebo och i andra med en känd effektivt läkemedel jämförelser, men slutsatsen är allmän. Eller inte beaktas arten av samtidig terapi;
- i fall av randomisering av dålig kvalitet (indelningen i grupper var inte tillräckligt slumpmässig).
Resultaten av metaanalyser hjälper läkaren att välja behandling, men de kan inte vara universella (för alla tillfällen) och kan inte ersätta läkarens kliniska erfarenhet.
Bevisnivåer
För att urskilja hur starkt en rekommendation kan lita på, nyanser (A, B, C) och nivåer av bevis (1, 2, 3, 4, 5). Jag tänkte ge denna klassificering här, men vid närmare granskning fick jag reda på att alla klassificeringar jag skiljer sig åt i detaljer, eftersom de antogs av olika organisationer. Av den anledningen ger jag bara ett av exemplen:
Detta är ett exempel på klassificering av bevisnivåer och graderingar av rekommendationer.
I ordning minskar trovärdigheten olika sorter studier arrangeras i följande ordning (källa - Swedish Council for Health Assessment Methodology):
- en randomiserad kontrollerad studie (dvs att ha en experiment- och kontrollgrupp med randomisering),
- icke-randomiserad kontrollerad studie med samtidig kontroll,
- icke-randomiserad kontrollerad studie med historisk kontroll,
- fallkontrollstudie,
- crossover kontrollerad studie, crossover studie,
- observationsresultat (öppen icke-randomiserad studie utan grupp),
- beskrivning av enskilda fall.
Hur man analyserar forskningsresultat
Alla resultat som erhållits i en klinisk prövning behandlas med metoder för matematisk statistik... Formlerna och principerna för beräkningar är ganska komplexa, en praktisk läkare behöver inte veta dem säkert, och i medicinska skolan vid den medicinska fakulteten presenteras de för dem i två fysiklektioner under det första året och används i socialhygien (hälsoorganisationer) under det sjätte året. Alla beräkningar utförs av arrangörerna av kliniska prövningar oberoende med hjälp av statistiska programvarupaket.
1) Statistisk tillförlitlighet. Inom medicin övervägs något värde statistiskt tillförlitligaom det bestäms med en sannolikhet på 95% eller mer. Detta gör att du kan utesluta oavsiktlig påverkan på det slutliga resultatet.
Om sannolikheten visade sig vara mindre än 95% är det nödvändigt att öka antalet analyserade fall. Om det inte hjälper att öka provet måste vi erkänna att det är svårt att uppnå ett tillförlitligt resultat i det här fallet.
2) Fel sannolikhet... Parameter betecknad med en latinsk bokstav sid (p-värde).
sid - sannolikheten för fel när man får ett tillförlitligt resultat. Det räknas i bråkdelar av en. För att konvertera till procent, multiplicera med 100. Det finns tre vanliga betydelser i kliniska studierapporter sid:
- p\u003e 0,05 - är INTE statiskt signifikant (det vill säga sannolikheten för fel är större än 5%),
- sid< 0.05 - является статистически значимым (вероятность ошибки 5% и меньше),
- sid< 0.01 - высокая статистическая значимость (вероятность ошибки не выше 1%).
Du kan nu förstå de flesta av resultaten i vetenskapliga tidskriftspublikationer. Öva:
En multicenter, randomiserad, prospektiv, öppen studie med blind slutpunktbedömning. ... En signifikant minskning av insulinkänslighetsindexet erhölls efter 16 veckors behandling jämfört med basvärden både i moxonidingruppen (p \u003d 0,02) och i metformingruppen (p \u003d 0,03). Det fanns inga signifikanta skillnader i denna indikator mellan studiegrupperna (p \u003d 0,92).
Kliniska studier av läkemedlet är ett nödvändigt steg i utvecklingen av något nytt läkemedel eller utvidgningen av indikationer för användning av ett läkemedel som läkare redan känner till. I de inledande stadierna av läkemedelsutveckling utförs kemiska, fysiska, biologiska, mikrobiologiska, farmakologiska, toxikologiska och andra studier på vävnader (in vitro) eller på laboratoriedjur. Dessa är de så kallade prekliniska studiervars syfte är att erhålla vetenskapliga utvärderingar och bevis för läkemedlets effektivitet och säkerhet. Dessa studier kan dock inte ge tillförlitlig information om hur de studerade läkemedlen kommer att fungera hos människor, eftersom organismen hos försöksdjur skiljer sig från människor både vad gäller farmakokinetiska egenskaper och vad organ och system svarar på läkemedel. Därför är det nödvändigt att genomföra kliniska prövningar av läkemedel på människor.
Så vad är det? klinisk studie (studie) av ett läkemedel? Detta är en systematisk studie av ett läkemedel genom användning hos en person (patient eller friska frivilliga) för att bedöma dess säkerhet och / eller effekt, samt för att identifiera och / eller bekräfta dess kliniska, farmakologiska, farmakodynamiska egenskaper, för att bedöma absorption, distribution, metabolism, utsöndring och / eller interaktioner med andra läkemedel. Beslutet att starta en klinisk prövning fattas av Sponsor / kundsom ansvarar för att organisera, övervaka och / eller finansiera forskningen. Ansvaret för det praktiska genomförandet av forskningen ligger i Forskare (person eller grupp av personer). Som regel är sponsorn läkemedelsföretag som utvecklar läkemedel, men forskaren kan också fungera som sponsor om forskningen inleds på hans initiativ och han är fullt ansvarig för dess uppförande.
Kliniska prövningar måste utföras i enlighet med de grundläggande etiska principerna i Helsingforsdeklarationen, GCP-reglerna ( Bra klinisk praxis, Good Clinical Practice) och tillämpliga lagkrav. Innan en klinisk studie inleds bör en bedömning göras av förhållandet mellan den förutsebara risken och den förväntade nyttan för ämnet och samhället. I spetsen ligger principen om prioritet för ämnets rättigheter, säkerhet och hälsa framför vetenskapens och samhällets intressen. Ämnet kan inkluderas i studien endast på grundval av frivilligt informerat samtycke (IS) erhållits efter en detaljerad bekantskap med forskningsmaterialet.
Klinisk forskning måste vara vetenskapligt baserad, detaljerad och tydligt beskriven i forskningsprotokoll... Det är ansvaret att utvärdera risk-nyttoförhållandet och granska och godkänna studieprotokollet och annan dokumentation relaterad till genomförandet av kliniska prövningar. Organisationens expertråd / Oberoende etikkommitté (ESP / NEC). Efter godkännande från ESP / IEC kan den kliniska prövningen påbörjas.
Typer av kliniska prövningar
Pilot studie syftar till att erhålla preliminära data som är viktiga för planering av ytterligare etapper av studien (bestämma möjligheten att genomföra studien i ett större antal ämnen, urvalsstorleken i den framtida studien, erforderlig forskningskraft etc.).
Slumpmässig klinisk prövningdär patienter slumpmässigt delas in i behandlingsgrupper (randomiseringsprocedur) och har samma möjlighet att få studien eller kontrollläkemedlet (jämförande läkemedel eller placebo). I en icke-randomiserad studie utförs inte randomiseringsförfarandet.
Kontrollerade (används ibland som en synonym "jämförande") en klinisk prövning där ett undersökande läkemedel, vars effekt och säkerhet ännu inte har studerats fullständigt, jämförs med ett läkemedel vars effektivitet och säkerhet är välkänd (komparatorläkemedel). Detta kan vara placebo, standardbehandling eller ingen behandling alls. PÅ okontrollerad (icke-jämförande) studie används inte kontroll- / jämförelsesgruppen (gruppen av försökspersoner som tar jämförelsemedicin). I bredare mening avser kontrollerad forskning all forskning där potentiella källor till partiskhet kontrolleras (om möjligt, minimerad eller utesluten) (dvs. den utförs i strikt överensstämmelse med protokollet, övervakas etc.).
Vid ledning parallella studier försökspersoner i olika grupper får antingen endast studieläkemedlet eller bara komparatorn / placebo. PÅ tvärsnittsstudier varje patient får båda läkemedlen som jämförs, vanligtvis i en slumpmässig sekvens.
Forskning kan vara öppnanär alla deltagare i studien vet vilket läkemedel patienten får och blind (förtäckt) när en (enblind studie) eller flera parter i studien (dubbelblind, trippelblind eller fullblind studie) hålls i mörkret om fördelningen av patienter per behandlingsgrupp.
Prospektiv studie genomförs genom att dela upp deltagarna i grupper som kommer att få eller inte kommer att få utredningsmedicin innan resultatet inträffar. Till skillnad från honom, i retrospektiv (historisk) studie undersöker resultaten från tidigare kliniska prövningar, dvs. resultat inträffar innan studien påbörjas.
Beroende på antalet forskningscentra där forskning bedrivs i enlighet med ett enda protokoll, är forskningen enda centrum och multicenter... Om en studie genomförs i flera länder kallas den internationell.
PÅ parallell studie jämför två eller flera grupper av försökspersoner, varav en eller flera får studieläkemedlet och en grupp är kontrollgruppen. Vissa parallella studier jämför olika behandlingar utan att inkludera en kontrollgrupp. (Denna design kallas oberoende gruppdesign.)
Kohortstudie Är en observationsstudie där en utvald grupp människor (kohort) observeras över tiden. Resultat för ämnen i olika undergrupper av denna kohort, de som var eller inte (eller utsattes för varierande grad) läkemedelsbehandling jämförs. PÅ prospektiv kohortstudie kohorter komponerar i nuet och observerar dem i framtiden. PÅ retrospektiv (eller historisk) kohortstudie kohorten väljs från arkivregister och deras resultat spåras från det ögonblicket till nuet.
PÅ fallkontrollstudie (synonym: studie av liknande fall) att jämföra personer med en viss sjukdom eller utfall ("fall") med personer i samma befolkning som inte har sjukdomen eller som inte hade det givna resultatet ("kontroll") för att identifiera sambandet mellan resultatet och tidigare exponering för vissa risker, faktorer. I forskning fallserie flera individer observeras, vanligtvis får samma behandling utan att använda en kontrollgrupp. PÅ fallbeskrivning (synonymer: fall från praxis, fallhistoria, beskrivning av ett enskilt fall) behandling och resultat undersöks hos en person.
För närvarande ges företräde till en sådan design av kliniska prövningar av läkemedel, som ger de mest tillförlitliga uppgifterna, till exempel vid genomförande av potentiella kontrollerade jämförande randomiserade och företrädesvis dubbelblinda studier.
Nyligen har rollen för kliniska prövningar av läkemedel ökat i samband med införandet av evidensbaserade medicinprinciper i praktisk hälso- och sjukvård. Den främsta bland dem är att fatta specifika kliniska beslut för behandling av en patient baserat på väl beprövade vetenskapliga bevis som kan erhållas under väl utformade, kontrollerade kliniska prövningar.
I början utvecklar läkemedelstillverkningsföretaget läkemedlets kemiska och molekylära formel och bestämmer också formen för dess frisättning (tablett, injektion, suspension, etc.).
När läkemedlet har skapats måste läkemedelsföretaget genomföra prekliniska studier av läkemedlet... Prekliniska studier inkluderar en mängd biologiska, mikrobiologiska, farmakologiska, kemiska, fysiska och toxikologiska studier på isolerade humana vävnader ( in vitro - "in vitro") eller försöksdjur ( in vivo). Huvudsyftet med preklinisk forskning är att skaffa data och bevis om effektiviteten och säkerheten för det undersökande läkemedlet. Men med hjälp av prekliniska studier är det omöjligt att förstå hur läkemedlet kommer att fungera i människokroppen, eftersom laboratoriedjurens kropp skiljer sig mycket från människans kropp. Det är därför, efter att ha genomfört en preklinisk studie av läkemedlet, är det nödvändigt att kontrollera dess effekt på människor - detta kommer att hända i minst 3 steg. Läkemedlet kommer att gå till varje nästa fas endast om det visar goda resultat i den nuvarande.
Fas I
Fas I-kliniska prövningar syftar till att bestämma tolerabilitet, preliminär säkerhetsbedömning och bestämma de farmakokinetiska och farmakodynamiska parametrarna för studieläkemedlet.
Fas I-kliniska prövningar involverar ett relativt litet antal volontärer, vanligtvis högst 100 personer. När man forskar mot cancerläkemedel rekryteras volontärer med lämpliga onkologiska sjukdomar... Fas I kliniska prövningar utförs på specialiserade institutioner som har nödvändig utrustning, till exempel intensivvård. Fas I-studier kan randomiseras och blindas.
Fas I kliniska prövningar undersöker läkemedelsabsorption, toxicitet, distribution, metabolism och utsöndring i kroppen, liksom den föredragna användningsformen och den säkra dosnivån. Fas I kliniska prövningar varar från flera veckor till 1 år.


Fas I är uppdelad i två grupper:
- Kliniska studier av enstaka eskalerande doser (Enstaka stigande dosstudier, SAD). I denna studiegrupp får ett litet antal patienter en enda dos studieläkemedel under hela uppföljningsperioden. Om inga biverkningar upptäcks under observationsperioden och de erhållna uppgifterna motsvarar den förväntade säkerhetsnivån, ökas dosen av det undersökande läkemedlet. Nästa grupp deltagare får en ökad dos av läkemedlet. Administreringen av läkemedlet med en eskalering av doserna fortsätter tills biverkningar uppträder. Detta ögonblick kallas för att nå den maximalt tillåtna dosen.
- Kliniska studier av flera eskalerande doser (Multipla stigande dosstudier, MAD). I denna grupp av fas I-studier genomförs försök för att bättre förstå farmakokinetiken och farmakodynamiken för ett nytt läkemedel vid administrering flera gånger. Under studiens gång får patienterna en låg dos av läkemedlet upprepade gånger. Efter varje injektion tas blod och andra fysiologiska vätskor för att bedöma läkemedlets beteende efter att det kommer in i människokroppen.
Endast 16 av de 100 läkemedlen som gick in i fas 1 får FDA-godkännande och kommer att släppas ut på marknaden.
Fas II
Efter att läkare har studerat farmakokinetiken, farmakodynamiken och den preliminära säkerheten för undersökningsmedicin under fas I-prövningar inleder det sponsrande företaget nästa fas. Fas II kliniska prövningar utförs på en patientpopulation som väljs ut enligt strikta kriterier och numrerar cirka 100 till 1000 personer.
Huvudmålet för kliniska prövningar i andra fasen är att hitta den optimala doseringsnivån, samt att välja dosregimen för nästa, tredje fas. Doserna av läkemedlet som patienterna får i detta skede är vanligtvis lägre än de högsta doserna som deltagarna i den första fasen tog.
I kliniska fas II-studier måste det finnas en kontrollgrupp av patienter som inte skiljer sig i sammansättning och kvantitet från gruppen som får studieläkemedlet. Patienter i dessa två grupper bör vara jämförbara i kön, ålder och tidigare behandling. I detta fall jämförs studieläkemedlets effektivitet och tolerans antingen med placebo eller med en annan aktivt läkemedel, vilket är standarden vid behandling av sjukdomen, för närvaron av vilken huvudgruppen av ämnen valdes.
Fas II är uppdelad i fas IIA och fas IIB.
Fas IIA är en pilotstudie som är utformad för att bestämma nivån av läkemedelssäkerhet i utvalda grupper av patienter med en specifik sjukdom. Uppgifterna i den kliniska fas IIA-studien inkluderar att bestämma patienternas känslighet för olika doser av läkemedlet, beroende på administreringsfrekvensen.
Fas IIB - Dessa är reglerade kliniska prövningar, vars huvudsakliga uppgift är att bestämma den optimala doseringsnivån för läkemedlet för genomförande av fas III-studier.
I sällsynta fall kombineras fas I och II kliniska prövningar för att testa både läkemedelseffektivitet och säkerhet samtidigt.


Fas III
Fas III kliniska prövningarbrukar referera till randomiserade kontrollerade multicenterstudier som involverar en stor grupp patienter - från 1000 personer eller fler.
Fas 3 kliniska prövningar är utformade för att bekräfta säkerheten och effekten av det läkemedel som genomgår prövningar, bedömda i tidigare studier, och för att jämföra det med standardbehandling för en specifik cancer.
Också i detta skede effektiviteten av den terapeutiska effekten av studieläkemedlet studeras beroende på dess dos.
I fall där den kliniska prövningen av den tredje fasen är avslutad och den positiva effekten av behandlingen kvarstår fortsätter patienterna att få detta läkemedel så länge som deras remission varar.
Dessutom kan kliniska prövningar av den tredje fasen genomföras om det sponsrande läkemedelsföretaget vill utöka indikationerna för användning av något läkemedel. Studier av detta slag klassificeras ibland som fas IIIB.
Efter att ett läkemedelsföretag bekräftat effektiviteten och säkerheten för ett nytt läkemedel i fas III-studier, registreringsunderlaget för läkemedlet bildas, som beskriver metodiken och resultaten av prekliniska studier och tre faser av kliniska prövningar av läkemedlet. Beskriver också funktionerna i läkemedelsproduktionen, dess sammansättning och hållbarhet. Efter registrering skickas registreringsunderlaget till den auktoriserade vårdmyndigheten som registrerar nya läkemedel.
- Nytt läkemedel mer effektivän kända läkemedel med liknande verkan;
- Nytt läkemedel har bättre tolerans jämfört med redan kända läkemedel;
- Nytt läkemedel effektiv i fall där behandling med registrerade läkemedel är ineffektiv;
- Nytt läkemedel har en synergistisk effekt vid kombinationsterapiutan att öka toxiciteten;
- Nytt läkemedel ekonomiskt mer lönsamtän redan kända läkemedel;
- Nytt läkemedel lättare att användaän redan registrerade droger;
- Nytt läkemedel har en mer bekväm doseringsform än läkemedel som redan finns på marknaden.
Fas IV
Fas 4 kliniska prövningar kallas också forskning efter marknadsföring... De utförs efter registrering av läkemedlet och är faktiskt efter marknadsföring, deras genomförbarhet ligger i att få information för att optimera användningen av läkemedlet. Dessa studier kan krävas av både vårdgivaren och det sponsrande läkemedelsföretaget.
Fas IV syftar till att samla in ytterligare information om sådana parametrar som: tidpunkten för behandlingen, interaktionen mellan ett nytt läkemedel och andra läkemedel eller livsmedel, en analys av användningen hos patienter i olika åldersgrupper, ekonomiska indikatorer, långsiktiga behandlingsresultat, samt insamling av ytterligare data om läkemedlets säkerhet och effekt på exemplet med en stor befolkning under lång tid tid.
Om sällsynta men farliga biverkningar upptäcks under kliniska fas 4-studier kan läkemedlet avbrytas och användningen begränsas.
-
 Terbinafin tabletter: bruksanvisning
Terbinafin tabletter: bruksanvisning
-
 Symtom och behandling av pyelonefrit hos ett barn - manifestationer, diagnos, läkemedel och förebyggande Vilken typ av sjukdom är njurpyelonefrit hos barn?
Symtom och behandling av pyelonefrit hos ett barn - manifestationer, diagnos, läkemedel och förebyggande Vilken typ av sjukdom är njurpyelonefrit hos barn?
-
 Hemlagade masker för hårdensitet och skönhet - de bästa recepten
Hemlagade masker för hårdensitet och skönhet - de bästa recepten
-
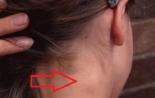 Infektiös mononukleos hos barn - symtom och behandling Mononukleosvirus hos barn är farligt
Infektiös mononukleos hos barn - symtom och behandling Mononukleosvirus hos barn är farligt